Childhood Eye Cancer — Parent’s Guide
Retinoblastoma treatment — eye-sparing options explained
Medically reviewed by Dr. C. Raghavendra Reddy, DM (Medical Oncology, Gold Medal) · Last reviewed June 2026
If your child has been diagnosed with retinoblastoma — or if you noticed a white glow in one eye in a photograph and a doctor has raised a concern — you deserve a clear, calm explanation of what this means and what modern treatment can offer. Retinoblastoma is a cancer of the retina that primarily affects young children. Today, a significant proportion of children can keep their eye with preserved vision, thanks to advances in eye-sparing therapy. This page explains how retinoblastoma develops, the warning signs, and the full range of treatment options available.
- Tumor board for every child — medical, surgical and radiation oncologists review the case together before any decision is made
- Eye-sparing treatment coordinated — our team coordinates with specialist ophthalmology and interventional radiology for intra-arterial chemotherapy when indicated
- 45-minute consultations — no rushed decisions; your questions about your child’s eye and vision are answered fully
- Free first consultation — paediatric oncology assessment at no charge, including written second opinion for families who need clarity on a report
17+
Cancer Specialists
on Panel
on Panel
96.9%
Breast Cancer
Survival Rate*
Survival Rate*
15,000+
Patients
Treated
Treated
4.8★
Google Rating
(800+ reviews)
(800+ reviews)
Understanding the diagnosis
What is retinoblastoma?
Retinoblastoma is a cancer that begins in the retina — the thin, light-sensitive tissue lining the back of the eye that receives images and sends them to the brain. It develops from immature retinal cells called retinoblasts that fail to mature normally during foetal or early childhood development, instead dividing without control and forming a tumour. Retinoblastoma is one of the few cancers where the genetic cause is well understood: in almost all cases it involves changes to a gene called RB1, which under normal circumstances keeps cell growth in check.
Retinoblastoma is almost exclusively a disease of young children. The great majority of diagnoses are made in children under five years of age, with many detected in infants. It can affect one eye (unilateral retinoblastoma, which accounts for roughly two thirds of cases) or both eyes simultaneously (bilateral retinoblastoma). When both eyes are involved, it is almost always the hereditary form of the disease. Bilateral retinoblastoma is typically diagnosed earlier — often in infancy — because family awareness or screening may be in place.
The hereditary form occurs when a child inherits one faulty copy of the RB1 gene from a parent who carries it. Because that first genetic change is already present in every cell of the body at birth, only one further change in a retinal cell is needed to trigger tumour development — which is why these children are at risk of tumours in both eyes and, in a small proportion, of certain other cancers elsewhere in the body later in life. In the non-hereditary (sporadic) form, both genetic changes occur by chance in a single retinal cell after birth; the child’s other cells are unaffected, and only one eye is at risk.
Retinoblastoma is not caused by anything the parents did during pregnancy. It is not contagious. There is no known environmental exposure, diet, or medication that causes it. Most families feel an instinct to search for what went wrong — but there is no fault to be found. The priority now is accurate diagnosis, classification of the tumour, and a treatment plan that gives your child the best chance of keeping their eye and their vision while ensuring the cancer is fully controlled.
Classification and warning signs
Forms of retinoblastoma and signs that prompt evaluation
Understanding whether retinoblastoma is unilateral or bilateral, hereditary or sporadic, and how advanced it is at diagnosis directly shapes the treatment path. Here is what each classification means in plain language.
One eye affected
Unilateral retinoblastoma
The tumour affects one eye only. Most unilateral retinoblastoma is the non-hereditary (sporadic) form, arising from two chance mutations in a single retinal cell after birth. It accounts for the majority of retinoblastoma cases. Children are typically diagnosed between the ages of one and three. Genetic testing is still offered, because a small proportion of unilateral cases are hereditary and carry the same long-term implications as bilateral disease.
- Most common presentation: white pupil reflex (leukocoria) or a new squint
- Tumour may be single or there may be multiple deposits in the same eye
- Genetic testing of the tumour tissue is recommended to guide family counselling
Both eyes affected
Bilateral retinoblastoma
Tumours develop in both eyes, almost always indicating the hereditary form. Bilateral retinoblastoma typically presents earlier in life — sometimes in the first months after birth — because the genetic predisposition means both eyes are at risk simultaneously. Each eye is staged and classified independently; treatment of the two eyes may differ because the tumour burden or tumour group may be different in each. Siblings and parents of a child with bilateral disease should be offered genetic screening.
- Almost universally hereditary (germline RB1 mutation)
- Each eye treated individually based on its own tumour group
- Long-term follow-up for second cancers is part of the care plan
Most common warning sign
Leukocoria — the white pupil reflex
In a flash photograph, a normal eye produces a red-eye effect because the flash reflects off the blood-vessel-rich retina. In an eye with retinoblastoma, the flash reflects off the white tumour surface, producing a pale or white glow in the pupil. This is called leukocoria. It is the first sign noticed in the largest proportion of children with retinoblastoma, and it is often noticed by a parent or family member in a photograph before any doctor visit. Leukocoria can also have other causes, but any white pupil reflex warrants urgent referral.
- Often visible in one or both eyes in photographs with flash
- May also appear as a white or yellow area visible to the naked eye in certain lighting
- Urgent ophthalmology assessment is required — do not wait for the next routine check
Second most common sign
Strabismus — a new squint
A squint (misalignment of the eyes, also called strabismus) is the second most common presenting sign of retinoblastoma, after leukocoria. It occurs when the tumour affects the part of the retina responsible for central (macular) vision, causing that eye to lose its ability to fix normally on an object. The brain then suppresses the input from that eye, and it drifts inward or outward. An important point: a squint in a young child always warrants an ophthalmology assessment, not just observation at home. While most squints in young children are not caused by cancer, retinoblastoma must be excluded.
- May be constant or intermittent at first
- Can appear as an inward or outward turn of one eye
- Should always prompt ophthalmology review, especially without a family history of squint
These signs do not mean your child definitely has retinoblastoma — each can have other causes. But because retinoblastoma responds best when caught early, any of these signs in a young child warrants prompt specialist assessment. — Explore the pediatric cancer hub · White glow in child’s eye — what to do
12+ Centres in Hyderabad · Pick yours
CION cancer care is closer than you think.
We're never more than 30 minutes away. Same panel of specialists at every centre. Same tumour board reviews. Same NCCN protocols. Pick the closest one and call directly — or let us pick for you.
Not sure which centre fits best? Tell us where you are — we'll suggest the closest one with the right specialists.
Help me pick the right centre
Beyond Hyderabad
35+ centres across Telangana & Andhra Pradesh
Travelling for treatment? We may have a centre right where you are.
Telangana
Andhra Pradesh
Don't see your city? Call 18002028726 — we'll find your nearest CION partner centre.
Meet the Specialists
17+ senior cancer specialists. One panel for your case.
Trained at AIIMS, Tata Memorial, and leading international centres. Combined 150+ years of experience. Every complex case is reviewed by 3+ of them — together.

Medical Oncologist

Medical Oncologist
Dr. C. Raghavendra Reddy
MBBS(Gold Medal), DNB(General Medicine), DM(Medical Oncology)(Gold Medal)

Medical Oncologist
Dr. Bharati Devi Gorantla
MBBS, MD(General Medicine), DM(Medical Oncology)(Adyar,Chennai), ECMO, MRCP SCE(UK)

Medical Oncologist
Dr. Owais Mohammed
MBBS, MD (General Medicine), DrNB (Medical Oncology), ECMO, MRCP SCE (Medical Oncology) (UK)

Medical Oncologist

Medical Oncologist

Surgical Oncologist
Dr. Muralidhar Muddusetty
MBBS (AIIMS), MS (Surgery) (AIIMS), DNB (Surgical Oncology), MRCS (Edinburgh)

Surgical Oncologist

Surgical Oncologist

Surgical Oncologist
Dr. Vinay Mamidala
MBBS, MS(General Surgery), M.Ch(Surgical Oncology), FMAS, FARIS(Ongoing)

Surgical Oncologist

Radiation Oncologist

Radiation Oncologist

Radiation Oncologist

Hematologist

Interventional Radiologist
Dr. Mohammed Imran

Surgical Oncologist
Dr. Vajja Sandeep Kumar
MBBS, MS (General Surgery), DrNB (Surgical Oncology), FALS Oncology

Surgical Oncologist
Want a specific doctor for your case? Mention them when booking.
Book Free ConsultationBook an appointment with our specialist
Share your name and number — we'll call you back within 30 minutes to schedule your consultation.
Every day counts with a retinoblastoma diagnosis
Our multidisciplinary team reviews every child’s case at a tumour board before any treatment begins. You deserve a second opinion from a coordinated team — not one specialist working in isolation.
Eye-sparing retinoblastoma therapy
How retinoblastoma treatment works — step by step
Modern retinoblastoma treatment is a coordinated process involving ophthalmology, paediatric oncology, interventional radiology, and pathology. Here is what the journey typically looks like, from the first specialist assessment to the final treatment decision and follow-up.
Examination under anaesthesia (EUA) and tumour classification
Because young children cannot cooperate with a detailed eye examination while awake, the first formal assessment of retinoblastoma is carried out under general anaesthesia. This examination under anaesthesia (EUA) allows the ophthalmologist to examine the retina thoroughly using specialised instruments, map the number, size, and location of tumour deposits, and assess whether the tumour involves the optic nerve or extends into structures beyond the eye. The findings are used to classify each affected eye using the International Classification of Retinoblastoma (ICRB), which assigns a group from A to E: group A represents small, peripheral tumours far from critical structures; group E represents very advanced disease with a high probability that the eye cannot be saved. This classification directly determines which treatment options are available.
Staging investigations — checking for spread beyond the eye
While retinoblastoma that is confined to the eye (intraocular) is highly treatable, any extension beyond the eye requires additional treatment planning. MRI of the brain and orbits is routinely performed to assess whether the tumour has reached the optic nerve, the orbit, or the brain. A lumbar puncture (spinal tap) and bone marrow biopsy may be performed if there is concern about spread. In practice, the large majority of retinoblastoma cases in children are diagnosed while still intraocular — but this assessment is essential to confirm that and to guide the overall treatment approach.
Intravenous chemotherapy — shrinking tumours to enable eye-sparing focal treatment
For eyes classified as group B, C, or D, intravenous chemotherapy (chemoreduction) is often the first treatment used. Chemotherapy medicines are given through a vein and travel through the bloodstream to reach the eye. The goal is to shrink the tumour deposits enough that they can then be destroyed by focal (local) treatments applied directly to the retinal surface. Chemoreduction is given in cycles over several months, with EUA performed at each cycle to monitor the response. Once the tumours have responded sufficiently, the treating team applies focal therapies — laser photocoagulation, cryotherapy, or a combination — directly to the remaining tumour during EUA. This two-phase approach — systemic chemotherapy followed by focal treatment — has enabled eye preservation in many children who previously would have required enucleation.
Intra-arterial chemotherapy — targeted delivery directly to the eye
Intra-arterial chemotherapy (IAC) — also called ophthalmic artery chemosurgery — has transformed eye preservation rates over the past fifteen years and is now standard at specialist retinoblastoma centres worldwide. A paediatric interventional radiologist guides a very fine catheter through an artery in the groin all the way to the ophthalmic artery, the blood vessel that supplies the eye. Chemotherapy medicine is then infused precisely at the point where it enters the eye. Because the medicine is delivered directly to the tumour rather than travelling through the whole body, the concentration in the eye is far higher than what systemic chemotherapy could safely achieve, while the exposure to the rest of the body is substantially reduced. IAC has enabled preservation of eyes previously considered unsalvageable, including some group D eyes. The procedure is performed under general anaesthesia and typically given in three to six sessions, each assessed by EUA. Suitability for IAC depends on tumour group, the anatomy of the blood vessels, and whether disease has extended outside the eye — the multidisciplinary team will assess this carefully.
Focal treatments — laser and cryotherapy for small tumour deposits
For small tumours, or for residual tumour deposits after chemoreduction, focal treatments applied directly to the retinal surface are effective and preserve surrounding healthy retinal tissue. Laser photocoagulation uses a focused beam of light to heat and destroy the tumour and its blood supply. Transpupillary thermotherapy (TTT) uses a lower-intensity infrared laser to heat the tumour more slowly over a longer duration, which can be effective for certain tumour types. Cryotherapy applies a freezing probe to the outer surface of the eye over the tumour to freeze and destroy it from the outside. These procedures are performed during EUA while the child is under general anaesthesia. They may be used alone for very small, anteriorly placed tumours, or in combination with chemotherapy as consolidation after tumour shrinkage.
Enucleation — when eye removal is the safest path
When the tumour is classified as group E — very advanced, occupying a large proportion of the eye — or when there are features suggesting the cancer has reached the optic nerve or is at risk of spreading beyond the eye, enucleation (surgical removal of the eye) is recommended. Enucleation is the most effective treatment for advanced intraocular disease and eliminates the primary source of cancer in a single procedure. The surgery removes the eyeball, preserving the surrounding orbital muscles and tissue so that a prosthetic eye can be fitted later. Children adapt remarkably well to monocular vision. A prosthetic ocular implant is placed at the time of surgery or soon after; an external prosthetic eye matched to the child’s eye colour is fitted once the socket has healed, usually within four to six weeks. Many children with a prosthetic eye attend school, play sport, and live full lives that are indistinguishable from those of their peers. After enucleation, the removed eye is examined by a pathologist to look for high-risk features that might indicate adjuvant chemotherapy or radiation is needed to protect against spread.
Genetic testing and long-term follow-up
After initial treatment, genetic testing is offered to identify whether the child carries a hereditary RB1 mutation. This has implications not only for the child — who will need long-term surveillance for second tumours if the hereditary form is confirmed — but for the entire family. Siblings of a child with bilateral retinoblastoma, or with confirmed hereditary unilateral disease, should be screened by an ophthalmologist as soon as possible after diagnosis, and at regular intervals during early childhood. The child’s follow-up after treatment includes regular EUAs at decreasing intervals, transitioning to awake slit-lamp examinations as the child grows older and can cooperate. Long-term follow-up with the oncology team continues into adulthood for children with the hereditary form, to monitor for any second cancers. CION’s multidisciplinary team coordinates this entire follow-up pathway, ensuring no step is missed.
Have questions about your child’s treatment options?
Our paediatric oncology team will review your child’s reports and explain which eye-sparing options may be appropriate. Free first consultation.
Coordinated Care
You don’t have to navigate this alone
Every family facing a retinoblastoma diagnosis deserves a team that is working together — oncologists, ophthalmologists, and support specialists — so no detail is missed and no decision is rushed. We walk this journey with you.
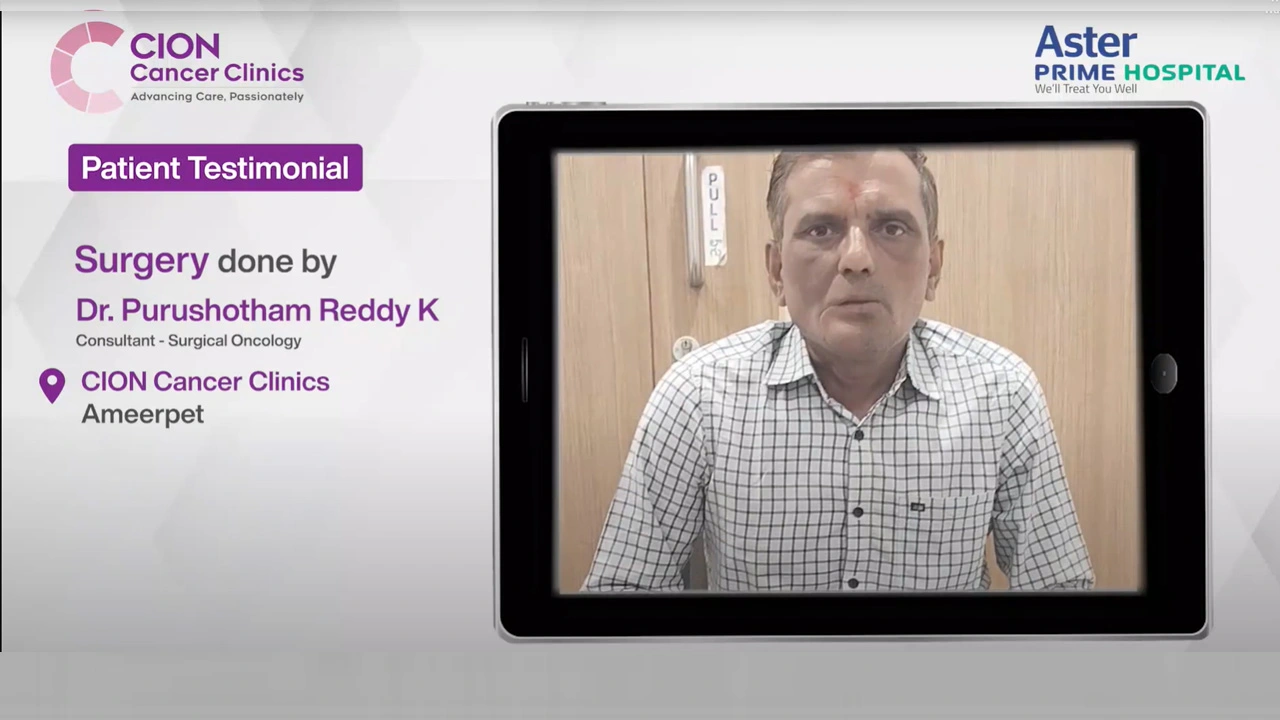
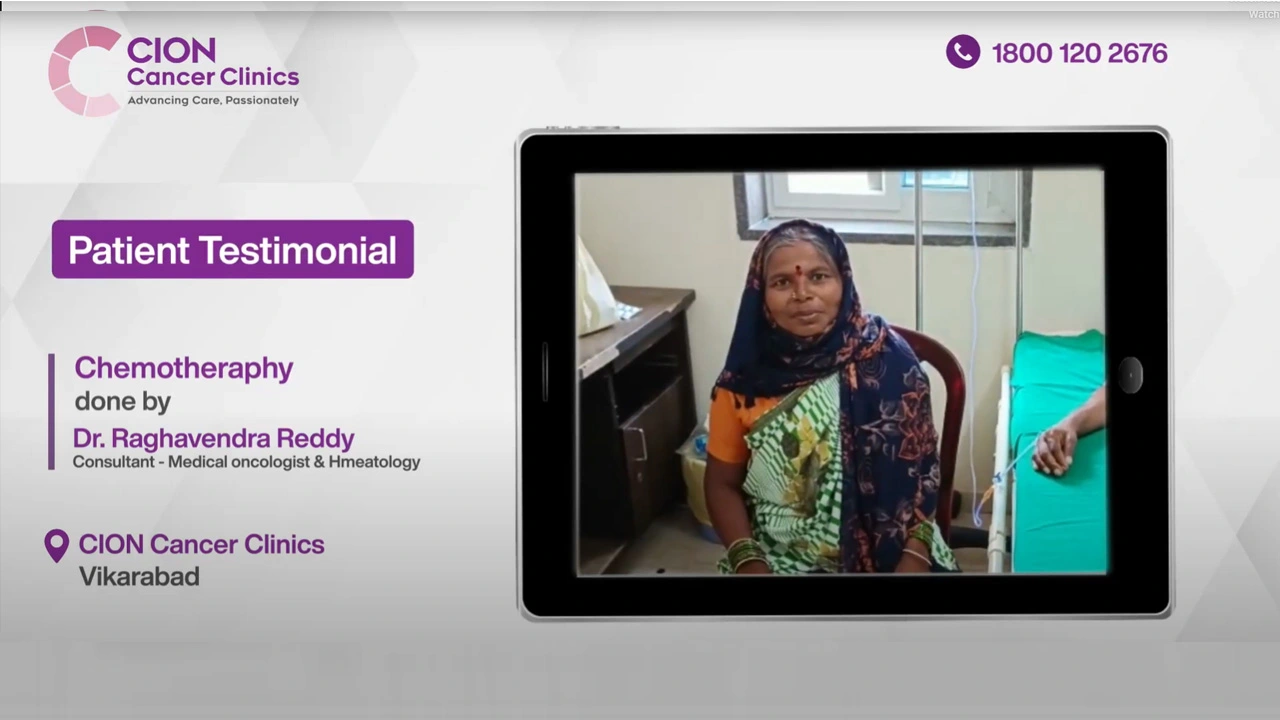
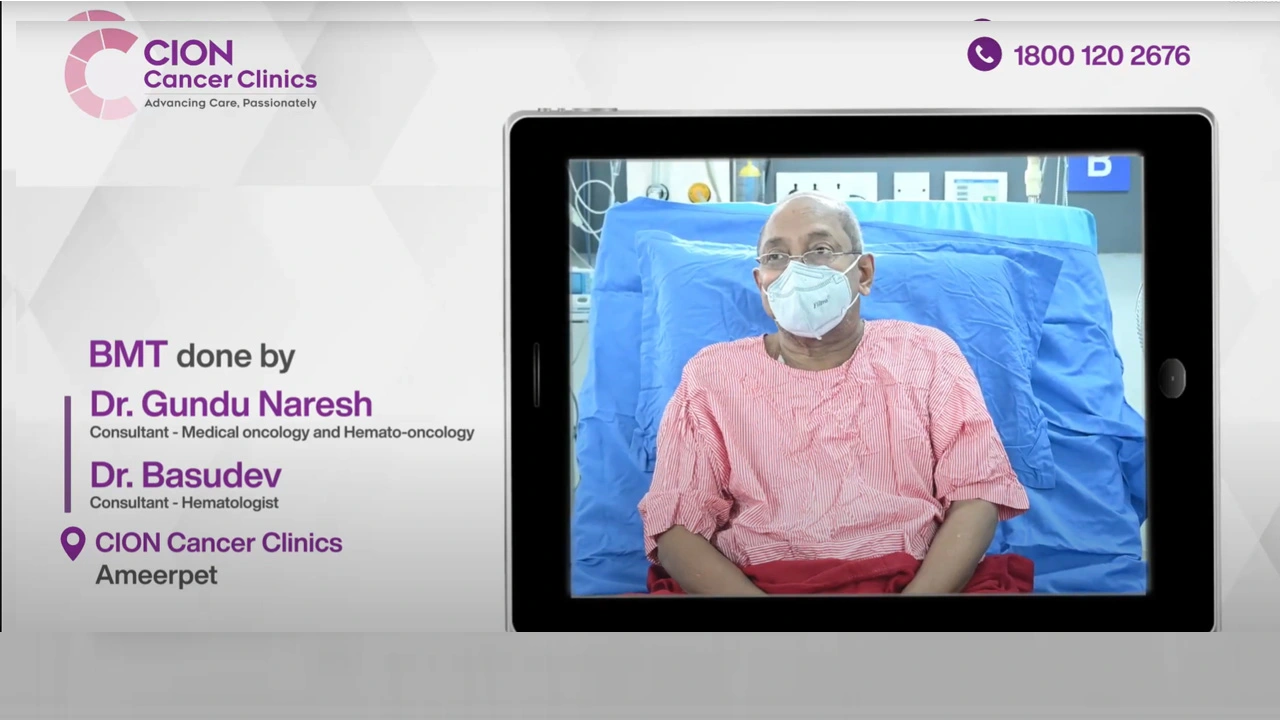
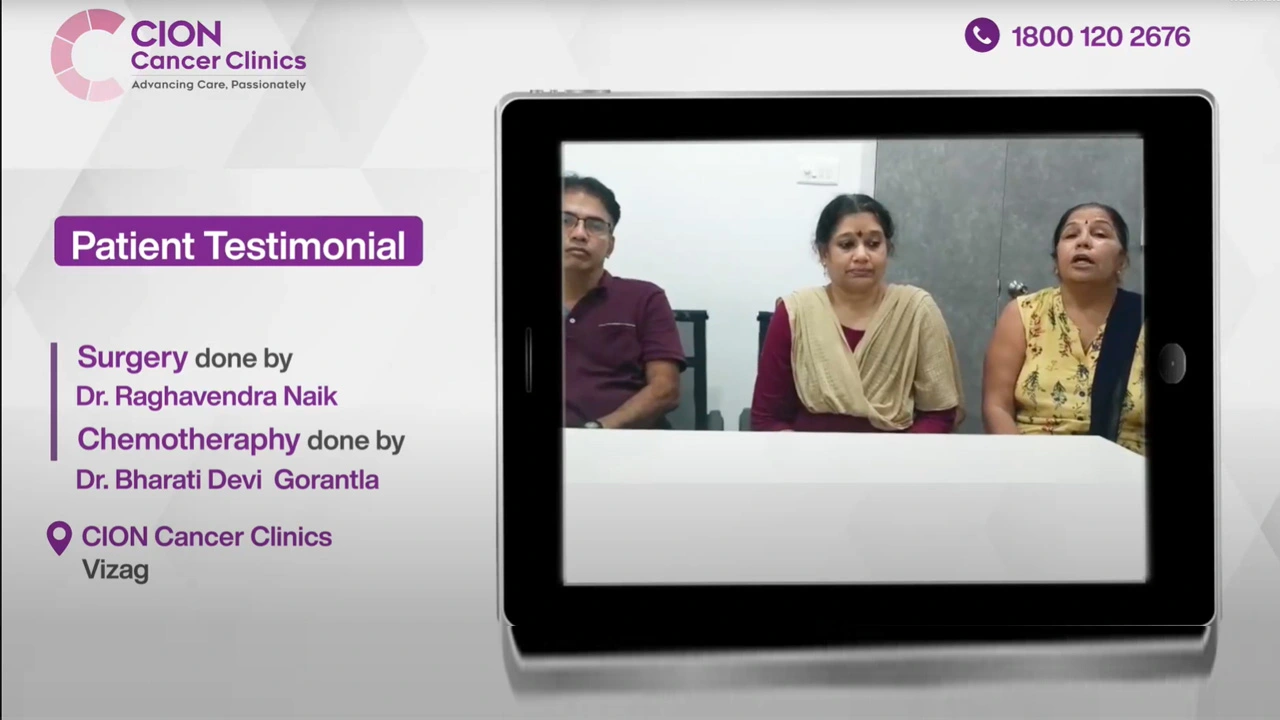
Real Stories. Real Voices.
15,000+ patients chose CION. Hear from them directly.
These aren't paid endorsements or written reviews. These are video testimonials from real patients and families — recorded on their own phones, in their own words. Pick any one. Watch it. Then decide.
4.8★800+ Google reviews
50+video testimonials
15,000+patients treated

















Read all 800+ reviews on Google
Start Your Story. Book Free Consultation.
Common questions
Your questions about retinoblastoma treatment — answered
What is retinoblastoma and which children does it affect?
Retinoblastoma is a cancer that begins in the retina — the light-sensitive layer at the back of the eye that sends visual information to the brain. It is almost exclusively a cancer of young children; most diagnoses occur in children under five years of age, and a significant proportion are detected in infants under two. It can affect one eye (unilateral retinoblastoma) or both eyes simultaneously (bilateral retinoblastoma). The cancer arises when retinal cells that are still developing carry a specific genetic change — an alteration in a gene called RB1. In the hereditary form, children inherit a faulty copy of RB1 from a parent and are at risk of developing retinoblastoma in both eyes and, later in life, other cancers. In the non-hereditary form, the genetic change occurs spontaneously in a single retinal cell, and only one eye is typically affected. Retinoblastoma is not caused by anything the parents did during pregnancy.
What is the white glow in the eye (leukocoria) that people describe as a warning sign?
Leukocoria — the white pupillary reflex, sometimes called “cat’s eye reflex” or “white glow” — is the most common first sign of retinoblastoma that a parent notices. In a photograph taken with a flash, a normal eye reflects the flash as a red dot (the familiar “red-eye” effect). In an eye affected by retinoblastoma, the flash reflects off the white tumour surface instead of the normal red retinal blood vessels, producing a pale or white glow in the pupil. Parents often first notice this in family photographs or when light catches the child’s eye at a certain angle. Other warning signs include a new and persistent squint (strabismus) in a child with no previous squint history, a visibly red or painful eye, a change in the colour or appearance of the iris, or a child who appears not to be tracking objects normally with one eye. Leukocoria is not always cancer — several other conditions can cause a white pupil reflex — but any white pupillary reflex in a child warrants urgent assessment by an ophthalmologist.
What does eye-sparing treatment mean, and is it always possible?
Eye-sparing treatment means treating retinoblastoma in a way that preserves the eye and as much vision as possible, rather than removing the eye surgically. Modern retinoblastoma treatment has shifted strongly toward eye-sparing approaches over the past two decades, and a significant proportion of children — especially those with smaller or less-advanced tumours — can now keep their eye with preserved or functional vision. Eye-sparing options include intravenous chemotherapy to shrink tumours, intra-arterial chemotherapy delivered directly into the eye’s blood supply, laser therapy (photocoagulation) and cryotherapy (freezing) applied to small tumours on the retinal surface, and intravitreal chemotherapy injected directly into the vitreous cavity of the eye for certain intraocular disease. Whether eye-sparing treatment is possible depends on how advanced the tumour is at the time of diagnosis, how many tumour sites there are in the eye, whether the tumour has reached the optic nerve or extended beyond the eye, and the overall health of the child. The International Classification of Retinoblastoma (ICRB) groups tumours into groups A through E, from smallest and most treatable to most advanced. Eyes with very advanced disease (group E) often require removal to ensure the cancer does not spread. The decision is always made by a multidisciplinary team, weighing the chance of saving the eye against the safety of the child.
What is intra-arterial chemotherapy and how is it different from regular chemotherapy?
Intra-arterial chemotherapy (IAC) — sometimes called ophthalmic artery chemosurgery — is a technique that delivers chemotherapy medicine directly into the ophthalmic artery, the main blood vessel that supplies the eye. A thin, flexible tube (catheter) is guided through a large artery in the groin all the way up to the ophthalmic artery under imaging guidance, and the medicine is then infused precisely at the site where it is needed. Because the medicine goes directly to the eye rather than travelling through the whole bloodstream, higher concentrations can be delivered to the tumour with far less effect on the rest of the body than standard intravenous chemotherapy. IAC has significantly improved the ability to preserve eyes that previously would have required removal, and it is now a standard part of retinoblastoma care at specialist centres. Not every child is a candidate for IAC — suitability depends on tumour group, the size and anatomy of the blood vessels, and whether the tumour has extended outside the eye. The procedure requires general anaesthesia in children and is performed by an experienced interventional team.
Will my child need their eye removed, and what happens if they do?
Enucleation — the surgical removal of the affected eye — is recommended when the tumour is so advanced that eye-sparing treatment cannot be safely offered, or when the risk of the cancer spreading outside the eye outweighs the benefit of attempting to save it. This most often applies to eyes classified as group E under the International Classification of Retinoblastoma, or to eyes where the tumour has reached or crossed the optic nerve. While losing an eye is a profound experience for any family, it is important to understand that enucleation is curative for most children with disease that has not spread beyond the eye, and it completely removes the source of cancer. Children adapt remarkably well to monocular vision; the remaining eye compensates, and children attend school, play sport, and lead full lives. A prosthetic eye (ocular prosthesis) is fitted after the socket has healed — typically within four to six weeks of surgery. Modern prostheses are carefully matched in colour and move naturally with the socket muscles, making them visually indistinguishable in most situations. Regular follow-up with the oncology team continues after surgery, and the child will also need monitoring for the hereditary form of retinoblastoma if relevant.
Does the hereditary form of retinoblastoma mean other family members are at risk?
Yes. Hereditary retinoblastoma is caused by a change (mutation) in the RB1 gene that can be passed from parent to child. Children with the hereditary form have this change in every cell in their body, not only in the retinal cells. This has two important implications. First, siblings and other first-degree relatives of a child with bilateral retinoblastoma, or a child with unilateral retinoblastoma that is found to be hereditary on genetic testing, have a meaningful chance of also carrying the mutation and should be offered genetic testing and ophthalmic screening. Second, children who carry the RB1 mutation have a higher-than-average lifetime risk of developing other cancers, particularly certain sarcomas, later in life — which means long-term oncological follow-up is recommended even after retinoblastoma is successfully treated. Genetic testing (usually a blood test) can identify whether the RB1 mutation is present, and the results have significant implications for the whole family. A paediatric oncology team with genetic counselling support will guide the family through this process.
Pediatric Cancer A–Z
Explore All Pediatric Cancer Topics
Browse our complete library of parent-facing guides, grouped by topic — from warning signs and cancer types to diagnosis, treatment, side-effect care, survivorship and family support.