Childhood Cancer Types — Parent’s Guide
Rhabdomyosarcoma in children — a soft tissue sarcoma, explained
Medically reviewed by Dr. C. Raghavendra Reddy, DM (Medical Oncology, Gold Medal) · Last reviewed June 2026
If your child has been diagnosed with rhabdomyosarcoma — often called RMS — or if you are trying to understand what a report means, you deserve a clear and honest explanation. Rhabdomyosarcoma is a soft tissue sarcoma in children that begins in cells meant to become muscle. It is the most common soft tissue sarcoma in young people, and it can arise in several different locations in the body. This page explains what childhood rhabdomyosarcoma is, where it appears, what warning signs to look for, how it is diagnosed, and what treatment involves.
- Tumor board for every child — not one doctor’s opinion; a full multidisciplinary team reviews each case before any treatment decision
- 45-minute consultations — your questions are heard fully; we do not rush a decision about your child’s care
- Free first consultation — paediatric oncology assessment at no charge for first-time patients
- Transparent costs — written treatment plan and cost breakdown before anything begins; no hidden charges
17+
Cancer Specialists
on Panel
on Panel
96.9%
Breast Cancer
Survival Rate*
Survival Rate*
15,000+
Patients
Treated
Treated
4.8★
Google Rating
(800+ reviews)
(800+ reviews)
Understanding the diagnosis
What is rhabdomyosarcoma?
The name “rhabdomyosarcoma” comes from three Greek words meaning striped muscle, flesh, and tumour. It is a cancer that arises in primitive mesenchymal cells — the very early, unspecialised building-block cells that, in normal development, would go on to form skeletal muscle and other connective tissues. Because these primitive cells are found throughout the body during foetal development, rhabdomyosarcoma can appear in places where you would not expect a muscle tumour: the area around the eye, the nasal cavity, the middle ear, the bladder, the prostate, the vagina, and the bile ducts, as well as the more expected locations like the arms, legs, and trunk.
Rhabdomyosarcoma is the most common soft tissue sarcoma in children and adolescents. The majority of cases are diagnosed in children under the age of ten, with a smaller second peak in teenagers. It is rare in adults. The cancer can be localised (confined to the original site), regionally spread (involving nearby lymph nodes), or metastatic (spread to distant organs such as the lungs, bone marrow, or bones) at the time of diagnosis. The extent of disease at diagnosis is one of the most important factors in how the oncology team approaches treatment.
Most cases of childhood rhabdomyosarcoma arise without any known cause. A very small number are associated with rare genetic syndromes, such as Li-Fraumeni syndrome or neurofibromatosis type 1, but these are exceptions. In the large majority of children, RMS develops without any family history of cancer and without any environmental exposure the family could have controlled or predicted. Parents should know that nothing they did, ate, or were exposed to during pregnancy caused this.
Because RMS can arise in so many different locations and can look similar to other childhood tumours on imaging, accurate pathological diagnosis is essential. The pathologist must confirm not only that it is rhabdomyosarcoma, but also which subtype it is — because the subtype influences risk classification and treatment intensity. This is why specialist oncology review, not just a general paediatric opinion, is important from the outset.
Subtypes & locations
Types of rhabdomyosarcoma your child’s team may mention
Childhood rhabdomyosarcoma is not a single disease. Two main subtypes exist, and where the tumour is located also matters clinically. Understanding the vocabulary helps you follow your child’s team’s discussions and ask the right questions.
Most common subtype
Embryonal RMS (ERMS)
Embryonal rhabdomyosarcoma is the more common subtype, accounting for the majority of childhood RMS cases. It tends to occur in younger children and most often arises in the head and neck region (including the orbit around the eye) and the genitourinary tract. Under the microscope, the tumour cells resemble foetal muscle at about 6–8 weeks of development. ERMS generally responds well to the combination of chemotherapy, surgery, and radiation used in treatment protocols.
- More common in children under 5
- Often in the head, neck, or urinary tract
- Generally associated with a more favourable risk profile
Higher-risk subtype
Alveolar RMS (ARMS)
Alveolar rhabdomyosarcoma gets its name from its microscopic appearance, which resembles the air sacs (alveoli) of the lung. It accounts for a smaller proportion of childhood RMS but is biologically more aggressive. ARMS more frequently carries a specific chromosomal rearrangement involving the PAX3 or PAX7 gene fused with the FOXO1 gene. The presence of this gene fusion is an important prognostic marker and influences risk grouping. ARMS can arise in any location but is more commonly found in the trunk and extremities.
- Carries PAX3/PAX7–FOXO1 gene fusion in most cases
- More often in trunk, limbs, or perineal area
- Generally assigned to higher-risk treatment groups
Common location
Head & Neck RMS (including orbital)
The head and neck is the most common primary site for rhabdomyosarcoma in children. Orbital RMS — arising in the soft tissue around the eye — is a particularly important subgroup because it often presents with visible signs (proptosis, swelling near the eye) that can be detected early. Parameningeal sites (near the base of the skull and sinuses) require particularly careful evaluation because of their proximity to the brain and the potential for the tumour to grow into the meninges.
- Orbital tumours often detected early due to visible signs
- Parameningeal location requires specialist evaluation
- Surgery planned to preserve sight and facial function
Second most common location
Genitourinary RMS
Rhabdomyosarcoma arising in the urinary tract or genitourinary organs — including the bladder, prostate, vagina, and paratesticular area — is the second most common primary site. Bladder and prostate tumours may present with blood in the urine, difficulty urinating, or a pelvic mass. Paratesticular RMS presents as a painless scrotal swelling and is important to distinguish from other scrotal conditions. Treatment planning in this location places particular emphasis on preserving bladder and reproductive function wherever possible.
- Blood in urine or urinary difficulty are key signs
- Paratesticular RMS presents as scrotal swelling
- Treatment aims to preserve urinary and reproductive function
Also common
Extremity & Trunk RMS
Rhabdomyosarcoma arising in the arms, legs, chest wall, or abdominal wall presents most often as a firm swelling or growing lump. These tumours may be painless initially, which is why parents sometimes assume the swelling is due to an injury. Extremity RMS is more likely to be of the alveolar subtype and has a higher likelihood of lymph node spread compared to other primary sites. The staging workup for extremity RMS always includes imaging of the regional lymph nodes, which may need to be sampled by biopsy or sentinel node procedure.
- Firm, growing lump that does not follow an injury
- Higher rate of lymph node spread than other sites
- Alveolar subtype more frequent in this location
Internal link: Explore all childhood cancer types at CION →
12+ Centres in Hyderabad · Pick yours
CION cancer care is closer than you think.
We're never more than 30 minutes away. Same panel of specialists at every centre. Same tumour board reviews. Same NCCN protocols. Pick the closest one and call directly — or let us pick for you.
Not sure which centre fits best? Tell us where you are — we'll suggest the closest one with the right specialists.
Help me pick the right centre
Beyond Hyderabad
35+ centres across Telangana & Andhra Pradesh
Travelling for treatment? We may have a centre right where you are.
Telangana
Andhra Pradesh
Don't see your city? Call 18002028726 — we'll find your nearest CION partner centre.
Meet the Specialists
17+ senior cancer specialists. One panel for your case.
Trained at AIIMS, Tata Memorial, and leading international centres. Combined 150+ years of experience. Every complex case is reviewed by 3+ of them — together.

Medical Oncologist

Medical Oncologist
Dr. C. Raghavendra Reddy
MBBS(Gold Medal), DNB(General Medicine), DM(Medical Oncology)(Gold Medal)

Medical Oncologist
Dr. Bharati Devi Gorantla
MBBS, MD(General Medicine), DM(Medical Oncology)(Adyar,Chennai), ECMO, MRCP SCE(UK)

Medical Oncologist
Dr. Owais Mohammed
MBBS, MD (General Medicine), DrNB (Medical Oncology), ECMO, MRCP SCE (Medical Oncology) (UK)

Medical Oncologist

Medical Oncologist

Surgical Oncologist
Dr. Muralidhar Muddusetty
MBBS (AIIMS), MS (Surgery) (AIIMS), DNB (Surgical Oncology), MRCS (Edinburgh)

Surgical Oncologist

Surgical Oncologist

Surgical Oncologist
Dr. Vinay Mamidala
MBBS, MS(General Surgery), M.Ch(Surgical Oncology), FMAS, FARIS(Ongoing)

Surgical Oncologist

Radiation Oncologist

Radiation Oncologist

Radiation Oncologist

Hematologist

Interventional Radiologist
Dr. Mohammed Imran

Surgical Oncologist
Dr. Vajja Sandeep Kumar
MBBS, MS (General Surgery), DrNB (Surgical Oncology), FALS Oncology

Surgical Oncologist
Want a specific doctor for your case? Mention them when booking.
Book Free ConsultationBook an appointment with our specialist
Share your name and number — we'll call you back within 30 minutes to schedule your consultation.
You deserve a specialist who has time for your questions
Every child with RMS is different. A 45-minute consultation with CION’s paediatric oncology team gives you a full picture — not a rushed summary.
The diagnostic journey
How childhood rhabdomyosarcoma is diagnosed
A thorough and accurate workup is essential before any treatment begins. At CION, no treatment decision is taken without the results of the full staging evaluation, reviewed at a multidisciplinary tumour board.
Clinical assessment and initial imaging
The oncology team begins with a detailed clinical history and physical examination. Initial imaging — typically an ultrasound of the affected area — gives a first look at the size, location, and characteristics of the mass. An MRI scan of the primary tumour site follows: MRI provides the most detailed picture of the tumour boundaries, its relationship to surrounding structures (blood vessels, nerves, adjacent organs), and helps the surgical team plan whether and how the tumour can be removed. Where MRI is not possible (for example, in very young children who cannot remain still), a CT scan is used as an alternative.
Chest CT and regional lymph node evaluation
A CT scan of the chest is essential in every child with suspected RMS to check whether the tumour has spread to the lungs, which is the most common site of distant spread. Imaging of the regional lymph nodes is also performed — either by CT, MRI, or PET-CT depending on availability and the child’s age. For some locations (particularly extremity and paratesticular RMS), regional lymph nodes must be evaluated surgically by sentinel node biopsy or surgical sampling, because imaging alone can miss small deposits of cancer in lymph nodes.
Biopsy and pathological analysis
A tissue biopsy is essential: a small piece of the tumour is removed by the surgeon and sent to the pathology laboratory. The pathologist examines the cells under a microscope and performs immunohistochemical staining and molecular tests — including fluorescence in situ hybridisation (FISH) or next-generation sequencing — to confirm the diagnosis of rhabdomyosarcoma, determine the subtype (embryonal or alveolar), and detect any PAX–FOXO1 gene fusion. The biopsy result is the single most important piece of diagnostic information and must be performed and reviewed by a pathologist with experience in paediatric tumours.
Bone marrow biopsy and bone imaging
Rhabdomyosarcoma can spread to the bone marrow and to bones. A bone marrow biopsy (taken from the hip bone under sedation or anaesthesia) checks whether the cancer has reached the marrow. Bone imaging — using either a bone scan (nuclear medicine) or a whole-body MRI — checks for spread to the skeleton. In centres where it is available, a PET-CT scan can provide whole-body staging information in a single session and is increasingly used in paediatric RMS staging. All bone marrow and bone imaging results are essential before the risk group is assigned.
Risk group assignment and tumour board review
Once all staging results are available, the multidisciplinary team meets at a tumour board to assign the child to a risk group (typically low, intermediate, or high risk). Risk grouping in RMS takes into account the primary tumour site, the extent of surgical removal if surgery was done first, whether the tumour has spread to lymph nodes, whether distant spread is present, the subtype (embryonal vs alveolar), and the molecular findings (PAX–FOXO1 gene fusion status). The risk group directly determines which treatment protocol the team recommends. At CION, we do not make this decision without a full team review.
Treatment overview
How rhabdomyosarcoma in children is treated
Treatment for childhood RMS always uses a combination of approaches. The exact combination depends on the risk group, the primary site, and how much of the tumour was removed at biopsy or initial surgery. At CION, care is led by a team — not one doctor acting alone.
Core of treatment
Chemotherapy
Systemic chemotherapy is a core component of treatment for virtually all children with rhabdomyosarcoma, regardless of risk group. It is given in cycles over a period of months. The goals of chemotherapy include shrinking the primary tumour before surgery (making complete removal more feasible), treating any cancer cells that may have spread beyond the primary site, and reducing the risk of relapse after local treatment. The number of cycles, the agents used, and the total duration depend on the risk group assigned by the tumour board.
- Used in all risk groups
- Given before and after surgery in most cases
- Helps treat cancer that cannot be seen on imaging
Local control
Surgery
Surgery aims to remove the primary tumour completely with clear margins — meaning no cancer cells remain at the edges of the removed tissue. Where complete removal is not possible without causing significant functional harm (for example, removal of the eye, or the bladder), the surgical team plans a limited resection after chemotherapy has shrunk the tumour as much as possible. Surgery is planned in close collaboration with the radiation oncologist, because the extent of surgical removal directly determines whether and how much radiation is needed afterwards.
- Aims for complete removal with clear margins
- Planned to preserve sight, continence, and limb function
- May follow chemotherapy to maximise resectability
Local control
Radiation Therapy
Radiation therapy is used to treat the primary tumour site when surgery has not achieved complete removal, or when the risk of local recurrence at the primary site is judged to be high. Modern radiation techniques allow the radiation oncologist to deliver a precise dose to the tumour while minimising exposure to surrounding healthy tissues — which is especially important in growing children to reduce the risk of long-term effects on bone growth, organ development, and secondary cancer risk. The radiation plan is prepared by a dedicated radiation oncologist as part of the tumour board.
- Used when surgical margins are not clear
- Precise modern techniques minimise dose to normal tissue
- Planned by a radiation oncologist specialising in paediatrics
Your child’s treatment plan starts with a conversation
Every child is different. A free 45-minute consultation lets you ask every question and understand every option before any decision is made.
Related: Bone cancer in children · Pediatric cancer treatment at CION
We walk this journey with you
You do not have to figure this out alone
Families at CION have a dedicated team — medical, surgical, and radiation oncologists, a nutritionist, and a psycho-oncologist — walking every step of the treatment journey with them.
Real Stories. Real Voices.
15,000+ patients chose CION. Hear from them directly.
These aren't paid endorsements or written reviews. These are video testimonials from real patients and families — recorded on their own phones, in their own words. Pick any one. Watch it. Then decide.
4.8★800+ Google reviews
50+video testimonials
15,000+patients treated















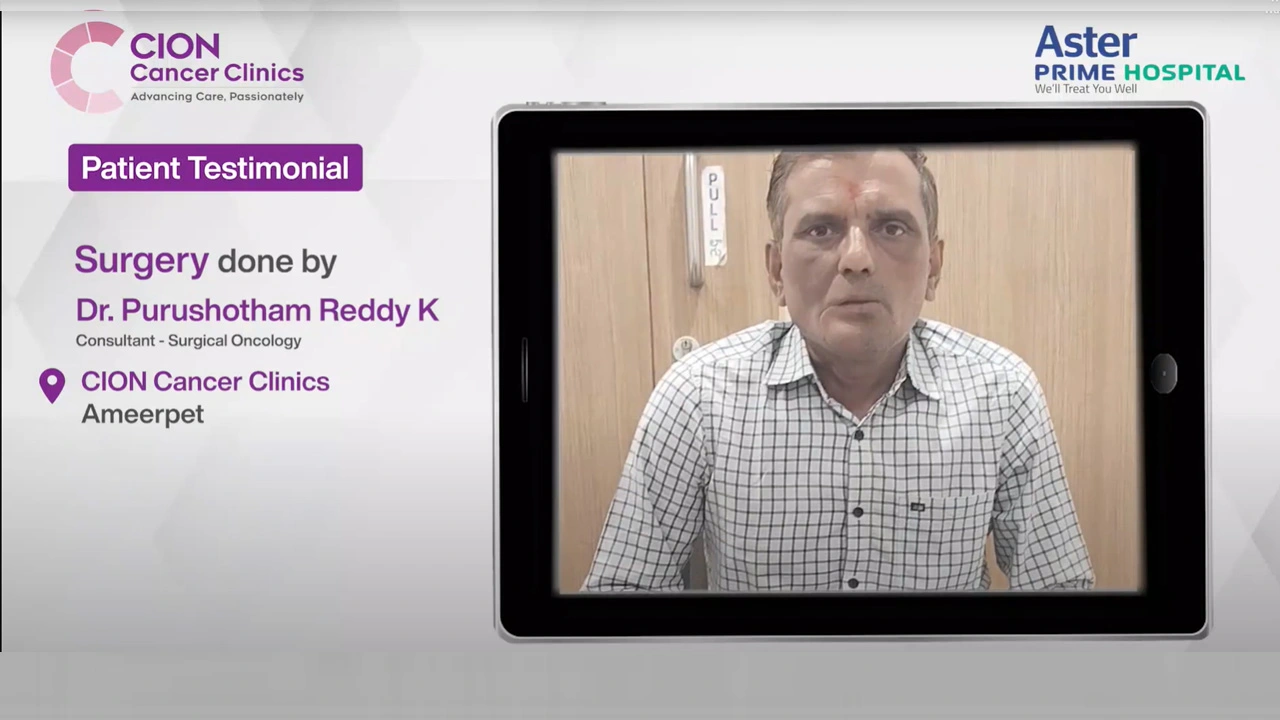
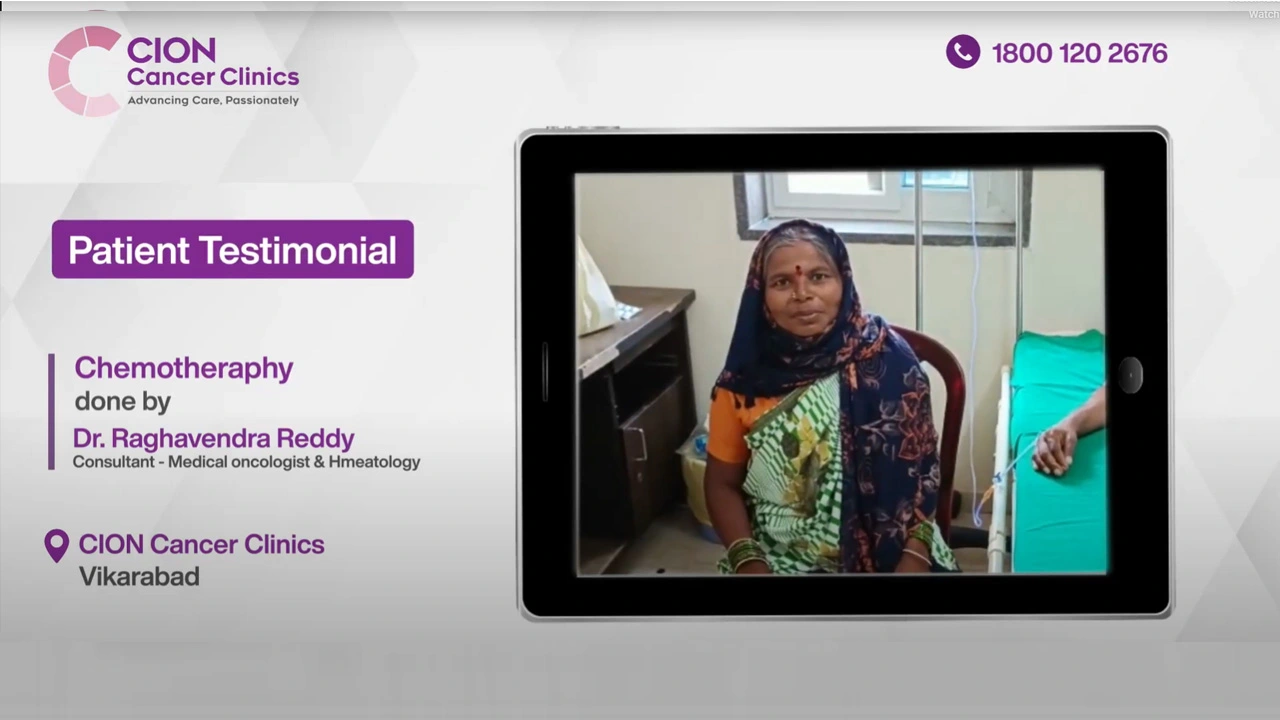

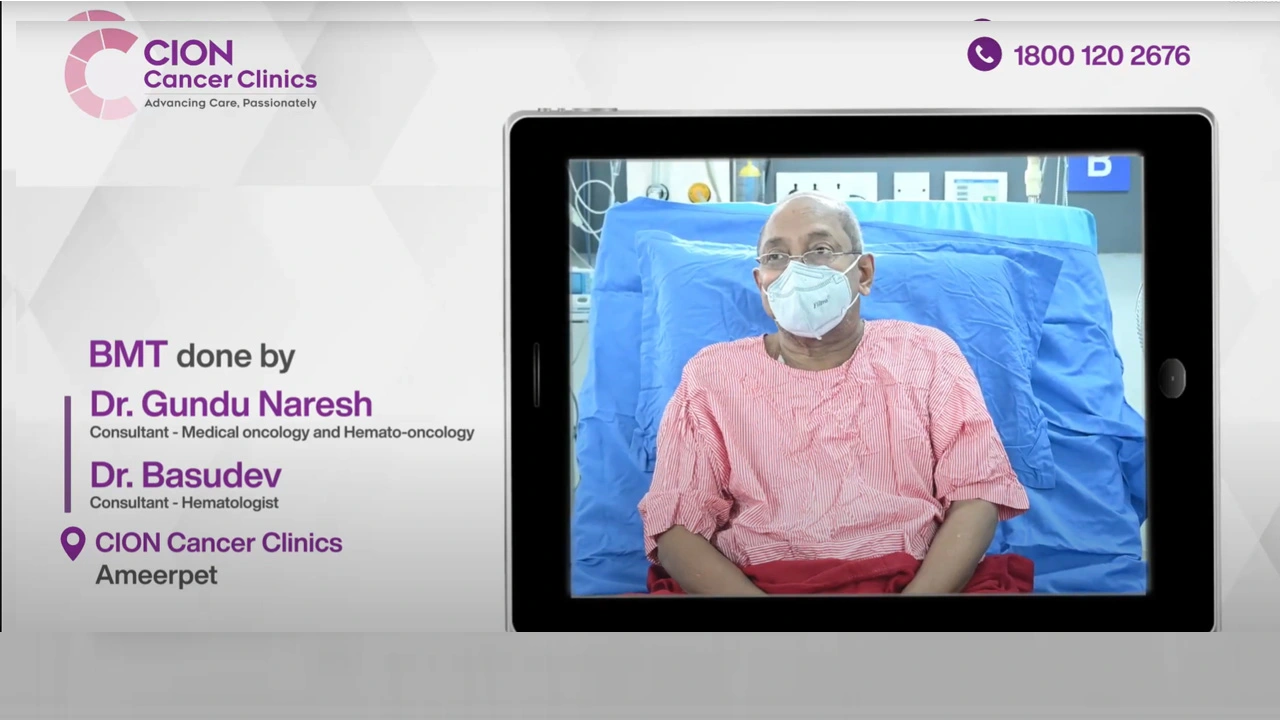
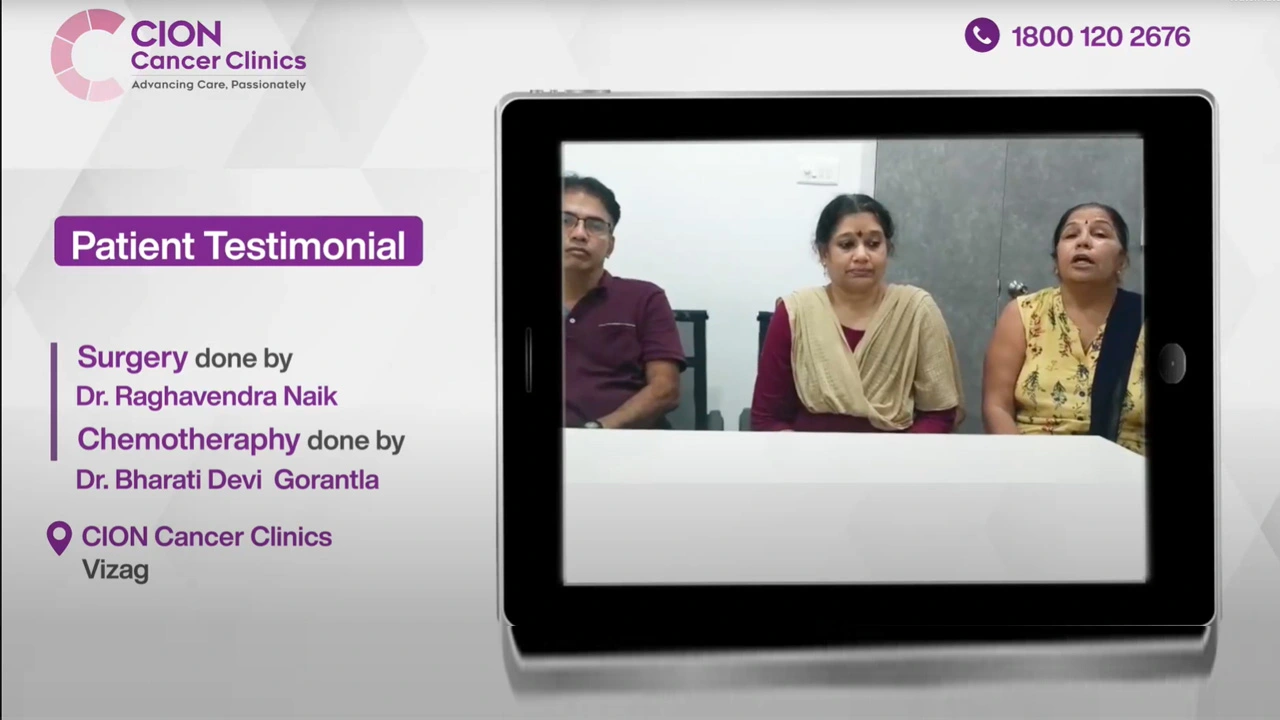

Read all 800+ reviews on Google
Start Your Story. Book Free Consultation.
Common questions
Your questions about rhabdomyosarcoma — answered
What is rhabdomyosarcoma and who does it affect?
Rhabdomyosarcoma (RMS) is a cancer that begins in cells that would normally develop into skeletal muscle. Despite its name, it can arise in parts of the body where skeletal muscle is not typically found — such as the head, neck, bladder, or the area around the eye (the orbit) — because the immature cells from which it develops are distributed widely in the body during foetal growth. RMS is the most common soft tissue sarcoma in children and teenagers, and the majority of cases occur in children under the age of ten. It is not caused by anything the family did; it arises from random changes in cell development. A very small number of cases are associated with rare inherited conditions, but most occur without any identifiable hereditary cause.
What are the warning signs of rhabdomyosarcoma in a child?
The warning signs of childhood rhabdomyosarcoma depend on where the tumour is growing. In the head and neck area, parents may notice a visible lump or swelling that does not go away, a bulging or protruding eye (proptosis), persistent blocked nose or nasal bleeding, a lump in or behind the ear, or drooping of the face. When the tumour involves the urinary tract or bladder, the child may have blood in the urine, difficulty passing urine, or a lump in the lower abdomen. Tumours in the limbs or trunk appear as a firm, painless (or occasionally painful) swelling or lump. Because the symptoms are location-dependent and can resemble much more common conditions — such as an injury or infection — any lump in a child that grows, does not resolve within two to three weeks, or is accompanied by other unexplained changes deserves a medical evaluation.
How is rhabdomyosarcoma diagnosed?
Diagnosing rhabdomyosarcoma involves imaging and tissue examination. The oncology team will typically start with an ultrasound of the affected area, followed by an MRI — which gives the most detailed picture of the tumour and its relationship to surrounding structures. A CT scan of the chest is done to check for spread to the lungs. A biopsy (removal of a small piece of tumour tissue) is essential: the pathologist examines the cells under a microscope and performs molecular and immunohistochemical tests to confirm RMS and determine the subtype, most importantly whether it is embryonal or alveolar. Bone marrow biopsy and imaging of the bones and lymph nodes are done to check for spread. All of these results together allow the multidisciplinary team to assign a risk group and plan treatment. At CION, every child's case goes to a tumour board before any treatment is finalised.
What are the two main types of rhabdomyosarcoma, and does the type matter?
The two main subtypes of rhabdomyosarcoma are embryonal (ERMS) and alveolar (ARMS). Embryonal RMS is the more common subtype and tends to occur in younger children; it is generally associated with a more favourable response to treatment compared to the alveolar subtype. Alveolar RMS — which accounts for a smaller proportion of cases — more frequently carries a specific chromosomal change (fusion of the PAX3 or PAX7 gene with FOXO1), and this is associated with a higher likelihood of spread and a more intensive treatment approach. The pathologist's report will specify which subtype is present and whether the relevant gene fusion is detected. This information is one of several factors used to assign a risk group, which directly determines the treatment intensity your child's team will recommend.
What does treatment for rhabdomyosarcoma in children involve?
Treatment for childhood rhabdomyosarcoma is always multimodal — meaning it uses more than one type of treatment. Chemotherapy is a core component for virtually all children with RMS, and it is used to shrink the tumour before surgery, to treat any spread in the body, and to prevent recurrence after local treatment. Surgery aims to remove the tumour while preserving as much function and appearance as possible — for example, preserving sight when the tumour is near the eye, or preserving bladder function when the tumour involves the urinary tract. Radiation therapy is used when surgery cannot achieve full removal or when the risk of local recurrence is high. The specific combination and sequence of treatments, and how many cycles of chemotherapy are given, depend on the risk group assigned to your child. Treatment is planned and delivered by a multidisciplinary team of medical, surgical, and radiation oncologists working together.
Can rhabdomyosarcoma spread to other parts of the body?
Yes, rhabdomyosarcoma can spread (metastasise) to other parts of the body, most commonly the lungs, bone marrow, and bones. The presence or absence of spread at the time of diagnosis is one of the most important factors used to determine the risk group and the intensity of treatment. Approximately one in five children diagnosed with RMS has evidence of spread to distant sites at the time of initial diagnosis. Children with localised disease — where the tumour is confined to the original site or nearby lymph nodes — tend to respond more favourably to treatment than those with widespread disease. This is why thorough staging investigations, including bone marrow biopsies and whole-body imaging, are an essential part of the initial workup before treatment begins.
Pediatric Cancer A–Z
Explore All Pediatric Cancer Topics
Browse our complete library of parent-facing guides, grouped by topic — from warning signs and cancer types to diagnosis, treatment, side-effect care, survivorship and family support.